RESEARCH/REVIEW ARTICLE
Measurement of loss rates of organic compounds in snow using in situ experiments and isotopically labelled compounds
Erika von Schneidemesser,1 James J. Schauer,1 Martin M. Shafer1
& Michael H. Bergin2
1 Environmental Chemistry and Technology Program, University of Wisconsin–Madison, 660 North Park Street, Madison, WI 53706, USA
2 School of Civil and Environmental Engineering and School of Earth and Atmospheric Sciences, 311 Ferst Drive, Georgia Institute of Technology, Atlanta, GA 30332, USA
Abstract
Organic molecular marker compounds are widely used to identify emissions from anthropogenic and biogenic air pollution sources in atmospheric samples and in deposition. Specific organic compounds have been detected in polar regions, but their fate after deposition to snow is poorly characterized. Within this context, a series of exposure experiments were carried out to observe the post-depositional processing of organic compounds under real-world conditions in snow on the surface of the Greenland Ice Sheet, at the Summit research station. Snow was prepared from water spiked with isotopically labelled organic compounds, representative of typical molecular marker compounds emitted from anthropogenic activities. Reaction rate constants and reaction order were determined based on a decrease in concentration to a stable, non-zero, threshold concentration. Fluoranthene-d10, docosane-d46, hexadecanoic acid-d31, docosanoic acid-d43 and azelaic acid-d14 were estimated to have first order loss rates within surface snow with reaction rate constants of 0.068, 0.040, 0.070, 0.067 and 0.047 h−1, respectively. No loss of heptadecane-d36 was observed. Overall, these results suggest that organic contaminants are archived in polar snow, although significant post-depositional losses of specific organic compounds occur. This has implications for the environmental fate of organic contaminants, as well as for ice-core studies that seek to use organic molecular markers to infer past atmospheric loadings, and source emissions.
Supplementary information to this article provides details on cleaning procedures and mathematical derivations of equations fit to the experimental data for observed losses and depositional flux. Please see the Supplementary Files, under Article Tools, online.
Keywords
Snow; photochemistry; air pollution; Greenland; Arctic.
Correspondence
James J. Schauer, Environmental Chemistry and Technology Program, University of Wisconsin–Madison, 660 North Park Street, Madison, WI 53706, USA.
E-mail: jjschauer@wisc.edu
(Published: 26 July 2012)
Polar Research 2012. © 2012 E. von Schneidemesser et al. This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License (http://creativecommons.org/licenses/by-nc/3.0/), permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Citation: Polar Research 2012, 31, 11597, http://dx.doi.org/10.3402/polar.v31i0.11597
The post-depositional fate of aerosol components in snowpacks, particularly in the polar regions, is the subject of increasing interest and study (Dubowski & Hoffmann 2000; Daly & Wania 2004; Herbert et al. 2006; Grannas, Jones et al. 2007 and references therein). Improved understanding of post-depositional processing for many organic species, including black carbon, is key to interpreting ice-core archives and snow pit records of past atmospheric aerosol loadings and changes in emission sources. In addition, the photochemistry and gas-phase exchange occurring within the snowpack and at the air–snow interface have a large impact on the oxidative capacity and overall chemistry occurring in the boundary layer above snowpacks (Domine & Shepson 2002; Klan & Holoubek 2002; Grannas, Jones et al. 2007). If incorporated into global atmospheric chemistry models, these dynamics would significantly improve the prediction capacity and accuracy of the models over polar regions. Clearly, there is a need for a better understanding of the post-depositional fate, specifically in terms of the photochemistry and other loss processes and loss rates of organic compounds in a snow-covered environment to be able to incorporate such parameters into the models (Hutterli et al. 2003; Daly & Wania 2004).
While the importance of photochemistry for many atmospheric compounds in polar regions is recognized, much of the recent focus has been on low molecular weight acids (Kawamura et al. 1995; Dibb & Arsenault 2002) and non-organic compounds, such as nitrogen oxides (Domine & Shepson 2002; Cotter et al. 2003; Jacobi et al. 2006). However, limited research has been conducted on the post-depositional processing of higher molecular weight organic compounds associated with atmospheric aerosols in snow and ice (Dubowski & Hoffmann 2000; Klan & Holoubek 2002; Klan et al. 2003; Dolinova et al. 2006). The carbon-13 or deuterium labelled compounds chosen for these experiments are based on their use as atmospheric tracer compounds in source identification. Alkanes and alkanoic acids are emitted from biogenic sources (e.g., abrasion from green and dead leaves), as well as anthropogenic sources (e.g., fossil fuel combustion sources such as vehicles or coal burning). The odd to even ratios of the alkane and alkanoic acid homologs and the presence of other species can be used to distinguish the contribution of these sources (Rogge et al. 1993). Polycyclic aromatic hydrocarbons (PAHs) are emitted from combustion sources, such as coal or biomass burning, with relative abundances of specific PAHs being used to distinguish between the sources (Oros & Simoneit 2000, 2001). If preserved after deposition to snow, these types of compounds could be used in the interpretation of ice-core records.
The few studies that have been conducted on the fate of the higher molecular weight organic compounds were typically carried out under laboratory conditions not necessarily representative of those in natural systems. The light source intensity used in some of these studies was much greater than that of natural light, while the wavelength range was not always representative of natural light conditions. Also, the concentrations of organic compounds in the snow samples were generally orders of magnitude greater than the concentrations observed in the Arctic or Antarctic (Dubowski & Hoffmann 2000; Jacobi et al. 2006; Grannas, Jones et al. 2007). However, evaluation of post-depositional processing at realistic trace level concentrations is experimentally quite challenging.
In this study, snow was prepared using a commercial snow-making machine with a water solution that contained stable, isotopically labelled compounds including n-alkanes, n-alkanoic acids, a low molecular weight diacid and a PAH. These compounds were chosen for their utility as molecular markers for sources and their presence in the snow and air at Summit, the research station at the peak of the Greenland Ice Sheet (Patton et al. 1991; Jaffrezo et al. 1994; Schauer & Cass 2000; von Schneidemesser et al. 2008; von Schneidemesser et al. 2009). They have long been used for source apportionment of urban aerosol samples (Schauer et al. 1996; Schauer & Cass 2000; Sheesley et al. 2007; Stone et al. 2008), and have great potential for furthering our knowledge of sources of air pollution transported to the Arctic. Understanding the degradation of these compounds is important to being able to utilize and accurately interpret records of these compounds in snow and ice. Instead of irradiating samples in a laboratory using an artificial light source, the samples were exposed on the surface of the Greenland Ice Sheet, at the Summit research station for periods of 24, 48 and 72 h. This paper examines the impact of environmental processes on the labelled compounds, as well as non-labelled compounds present in the contaminant-labelled snow over the time span of 72 h.
Methods
Isotopically labelled snow production
A water solution containing six deuterium labelled organic compounds (CDN Isotopes Inc, Pointe-Claire, QC, Canada; Sigma Aldrich, St. Louis, MO, USA; Cambridge Isotope Laboratories, Andover, MA, USA) was prepared as follows. The labelled compounds were first dissolved in either 100 mL of water (azelaic acid-d14), methanol (docosanoic acid-d43 and hexadecanoic acid-d31), methanol (fluoranthene-d10) or iso-octane (docosane-d46 and heptadecane-d36) to assure complete dissolution. A 0.1 mL aliquot of each of these solutions (0.02 mL for the solution with the PAH) was then spiked into a pre-cleaned 10-L carboy (HDPE) containing ≥18.0 Mohm Milli-Q water, and thoroughly mixed, so that the organic solvent present in the water was <0.1% (spiked concentrations are listed in Supplementary Table S1). This solution was then used to produce the snow.
A commercial snow-maker for home snow production was purchased from CHS Snowmakers (Littleton, CO, USA) The mainly aluminium body was fitted with Teflon lined hoses for air and water delivery. Air was delivered via an air compressor with a regulator to the snow-maker. The water solution was delivered using a peristaltic water pump, so that the water solution only passed through cleaned hoses. Prior to snow production, all snow-maker parts and associated hoses were thoroughly cleaned (see the Supplementary File for details).
Snow was produced in a walk-in freezer kept at approximately −21°C, in two batches on sequential days under the same conditions, and collected on new, clean plastic sheeting. The 10-L water solutions for each batch of snow (hereafter batch A and batch B) were made from the same 100 mL spike solutions. After production (30–45 min) the snow was collected in pre-cleaned 1-L glass bottles with Teflon-lined lids. Further details on all cleaning procedures can be found in the Supplementary File. The snow was stored in a freezer until transport, at which point the more than 80 1-L bottles were packed in coolers with ice packs that had been frozen at −80°C and transported via commercial airlines to Greenland. During stopovers (e.g., Kangerlussuaq, Greenland) the coolers were stored in walk-in freezers. Once at Summit, the bottles were stored outside, and in the coolers to avoid light exposure. The snow was produced approximately 6 weeks before use. For the return trip, the coolers were re-packed with fresh ice packs, and transported via military aircraft to New York, where the samples were loaded onto a freezer truck for transport to Madison, Wisconsin. The snow was then stored in standard freezers and remained frozen until analysis. Overall, no melting was observed in the majority of the samples. A few sample bottles did show signs of minimal melting near the edges of the bottles. The assumption was made that the concentrations observed in the control sets accurately represent t=0 h concentrations and that the compounds did not undergo any losses during transport or storage where they were kept frozen and not exposed to light. Furthermore, the assumption was that any non-labelled compounds that were quantified in the t=0 h and exposed samples were incorporated during the snow-making process.
Exposure experiments
At Summit, Greenland, a series of four exposure experiments were undertaken with the target compound-labelled snow. For exposure the snow was put in 0.56 m×0.66 m×0.10 m (approximate inner dimensions) pre-cleaned low-density polyethylene (LDPE) bins. For each experiment three bins for the three exposure lengths (24, 48, 72 h) were prepared by transferring snow from sample bottles into the bins. Each bin contained five bottles (1-L volume bottles) of snow. The snow transferred from the bottles into the bins for exposure resulted in a layer of snow in each bin of approximately 5 cm depth. The bin size and amount of snow was chosen so that a thin layer of snow would be exposed, and light exposure to all parts of the snow would be relatively even (e.g., the depth would not be significant enough that upper layers would shield lower layers of snow). The exposure was started at approximately the same time for all bins in the experiment. One additional set of five bottles, equivalent to the amount of snow that would have been transferred into one bin for an experiment, was left in the bottles and not exposed at all and represented the control. Each of the four experiments conducted had such a set of five bottles. These control samples were never removed from their jars for exposure but remained frozen in the coolers so that they were not exposed to light, the possibility of volatilization and/or wind stripping. One bin was collected at 24, 48 and 72 h each, returned to the 1-L bottles, and kept frozen and dark until analysis.
The LDPE bins containing the snow were set out on the snow surface at a satellite camp 1 km to the south of the main Summit research station camp. Due to drifting snow and wind, a wind fence was constructed around the bin exposure area 12 h after the exposure of the first experiment. The LDPE bins were weighed empty, and before and after exposure with the labelled-compound-containing snow. Changes in weight before and after exposure were minimal, on average within 4.6% of the initial weight. Any significant snow accumulation due to drifting (only relevant for the first experiment in the first 12 h of exposure), or snowfall was very carefully removed from the bins to avoid possible dilution. It should be noted that this was only necessary at certain points for experiments 1 and 2. Experiments 3 and 4 were not affected by significant amounts of fresh snowfall or drifting snow. Experiments 1 through 4 were set out on 14, 19, 21 and 22 July, respectively, typically in the early evening. During this time of year at Summit there was 24-h sunlight. Average daily temperatures during the experiments ranged from −19 to −3°C, with an overall average daily temperature of −11°C; maximum daily temperatures ranged from −13 to −2°C; minimum daily temperatures ranged from −29 to −9°C. Average daily wind speeds ranged from 0.9 to 3 m s−1, with an overall average for the whole time period of 1.8 m s−1. Maximum wind speeds reached 4.5 m s−1.
For consistency, since two batches of snow containing labelled compounds were produced, snow from batch A was used exclusively in experiments 1 and 3, and snow from batch B exclusively in experiment 2 (see Fig. 1, Supplementary Fig. S1). Since not enough snow remained from either batch for a complete fourth experiment, snow from both batches (A and B) was used in experiment 4 (batch A for the 24- and 72-h bin sets; batch B for the control [0 h] and 48-h bin sets). Four experiments were done to assure that any observed changes were reproducible and not due to uncontrollable factors, such as weather.

Fig. 1
Average concentration normalized to the concentration at time zero versus the exposure time. The lines are based on the determined reaction orders and reaction rate constants (see Table 1). Error bars represent the propagated error of the standard deviation of the blank values for the compound class (e.g., n-alkanes) as well as a percentage of the initial sample value.
Snow analysis
The chemical analysis method used is explained in more detail by von Schneidemesser et al. (2008). Here only a brief description will be given. The samples were melted in the glass bottles under a hood not more than 24 h before analysis. Two sample bottles (each 1-L in volume) were combined and analysed as one sample (approximately 700–1000 mL meltwater; snow density of 35–50%). Each meltwater sample was spiked with an internal standard, which included pyrene-d10, benz(a) anthracene-d12, coronene-d12, cholestane-d4, pentadecane-d32, eicosane-d42, tetracosane-d50, triacontane-d62, dotriacontane-d66, hexatriacontane-d74, decanoic acid-d19, tetradecanoic acid-d27, heptadecanoic acid-d33, eicosanoic acid-d39, tetracosanoic acid-d59 and levoglucosan-13C6, to assess analytical recovery, prior to the liquid–liquid extraction method using dichloromethane (DCM). The liquid–liquid extraction was performed by mixing the meltwater sample with DCM in a large separatory funnel for 2 min before collecting the DCM extracts, as detailed by von Schneidemesser et al. (2008). The DCM extracts were concentrated by rotary evaporation to approximately 1–2 mL and transferred to 2 mL vials for further volume reduction under a stream of high purity nitrogen. To ensure that all of the extract was transferred to the 2 mL vial from the round bottom flask, the flask was rinsed three times with approximately 1 mL aliquots of DCM that was then added to the vial. This procedure was repeated with an additional sample (again using two sample bottles). One of the sample extracts in a vial was then reduced to 200 µL and analysed by regular injection (3 µL) gas chromatography–mass spectrometry (GC–MS). The other sample extract was reduced to 100 µL and analysed by large volume injection (30 µL) programmable temperature vaporizer (PTV)–GC–MS. The large volume injection allows for enhanced sensitivity and lower detection limit for compounds present at lower concentrations, such as PAHs. The extract analysed by regular injection was first derivatized using diazomethane to convert the carboxylic acids to methyl esters (Schauer & Cass 2000). Labelled compounds were quantified using the calibration curve for the “identical” non-labelled compound (Sheesley et al. 2004; Stone et al. 2008). Average analytical recovery of the “identical” non-labelled compounds was 68%. Standard deviations for average recoveries were typically less than 10%. Concentrations were not corrected for recovery, but were blank subtracted. For individual percent recoveries for the method see von Schneidemesser et al. (2008). For blank values, see the Supplementary File. Using the two GC-MS analysis methods, non-labelled PAHs, alkanes and various acids were quantified, in addition to the isotopically labelled compounds listed in Table 1.
Table 1 Concentration of the labelled compounds in the water solution and the snow, the average retention from water solution to snow, reaction rate constants, reaction order and approximate half-life of the labelled compounds in snow.
| Compound |
Spike concentration in water (ng L−1) |
Avg snow concentrationa (ng L−1) |
Avg snow retentiona (%) |
Order |
Reaction rate constant (k) |
Normalized threshold value (C*/C0) |
Half-life (t1/2) (hours) |
r2
|
| Fluoranthene-d10
|
560 |
130±4.9 |
23±0.87 |
1 |
0.068±0.013 |
0.096b
|
10 |
0.93 |
| [C]=[C0]*e−0.068*t
|
|
110±4.2 |
20±0.75 |
|
|
|
|
|
| Heptadecane-d36
|
3100 |
240±10 |
7.8±0.33 |
n.a. |
0.00011±0.001 <0.003 |
n.a. |
n.a. |
0.0054 |
|
|
120±3 |
3.8±0.10 |
|
|
|
|
|
| [C]=[C0] – <0.003*t |
|
|
|
|
|
|
|
|
| Docosane-d46
|
2530 |
890±82 |
35±3.2 |
1 |
0.040±0.0029 |
0.60b
|
17 |
0.97 |
| [C]=[C0]*e−0.040*t
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
770±80 |
30±3.2 |
|
|
|
|
|
| Hexadecanoic acid-d31
|
2440 |
1190±77 |
49±3.1 |
1 |
0.070±0.0055 |
0.42 |
9.9 |
0.98 |
| [C]=[C0]*e−0.070*t
|
|
1480±31 |
61±1.3 |
|
|
|
|
|
| Docosanoic acid-d43
|
2430 |
1640±110 |
68±4.4 |
1 |
0.067±0.0038 |
0.58b
|
10 |
0.99 |
| [C]=[C0]*e−0.067*t
|
|
1040±630 |
43±26 |
|
|
|
|
|
| Azelaic acid-d14
|
1110 |
210±27 |
19±2.4 |
1 |
0.047±0.0012 |
0.37 |
15 |
0.997 |
| [C]=[C0]*e−0.047*t
|
|
210±40 |
19±3.6 |
|
|
|
|
|
| aThe concentrations are those measured in the control (no exposure) samples. The snow was produced in two batches and the values shown are separated into the concentrations quantified for batch A (top value) and batch B (bottom value). See the methods section for more detail. |
| bThreshold is an average of the 48- and 72-h values because the lowest value did not occur at 72 h. |
Results and discussion
Labelled organic compounds
The average concentrations and standard deviations of the labelled compound concentrations in the snow for the control sets (the isotopically labelled snow samples which were not exposed) are listed by batch in Table 1. Based on the spike concentration in the water solution, an average snow incorporation percentage was calculated, which reflects the amount of the labelled compounds measured in the snow relative to the possible concentration had the spiked water solution been 100% effectively translated into snow (Table 1). While these percentages may seem low, they are reproducible throughout the four sets. The losses may be due to loss during storage and transport of the snow, but most likely occurred during the snow-making process due to the necessary use of Teflon tubing, LDPE and HDPE containers. It is not likely that these losses were due to the analysis techniques, which were previously developed and tested (von Schneidemesser et al. 2008). No trends were observed for the percent recovery with any of the physical and chemical data that were investigated, including molecular weight, boiling point, melting point, water solubility, vapour pressure and octanol-water partitioning coefficient (see Supplementary Table S1). Even so, the carboxylic acids would be less volatile due to their higher molecular weights, hydrogen bonding with water molecules, and the possibility of deprotonation which would yield a non-volatile form of the acid. This may partially explain the higher snow retention rates of the hexadecanoic acid-d31 and docosanoic acid-d43. The percent recovery did not impact any of the experimental exposure results, as the starting point for the exposure experiments and the time series analysis was after the snow production. The average snow concentrations in the control sets (Table 1) are used as time zero (C0) values for the kinetic loss experiments. One of the control set concentrations, set 2 for docosanoic acid-d43, is significantly lower than the remaining three concentrations, which compare well (Supplementary Fig. S1). To avoid a bias in the calculated rate constants and other factors due to the anomalously low control (0 h) concentration, set 2 was omitted from the calculations.
In general, the concentrations for each exposure time were very similar for both batches and all experiments, when the uncertainty in the measurement (represented by the error bars) and the real-world conditions (daily variations in sunlight intensity, snow and/or fog deposition events, etc.) were considered (Fig. 1, Supplementary Fig. S1). The anomalous docosanoic acid-d43 in set 2 is an exception previously noted. Also, more variability in the conversion rate of heptadecane-d36 and azelaic acid-d14 was observed, which may be related to the lower initial concentration of these compounds in the labelled snow.
Linear regression analysis was performed on each of the experimental data sets for the isotopically labelled compounds to determine the order of their loss reactions. The model equations (see the Supplementary File for details) incorporated a steady-state concentration, or threshold value (C*), at which no further loss was observed; a model without a threshold value would not be sufficient to explain the data since 100% loss is not observed. We chose the threshold value (C*) to be the lowest concentration reached by each of the labelled compounds, typically at the 72-h exposure period. In the cases where the lowest concentration was not reached at 72 h, the average of the 48- and 72-h values was used. In these cases the 48- and 72-h values were statistically equivalent (e.g., were within the overlap of the error bars of the measurement). Slight statistically insignificant differences in these measurements represent analytical uncertainty and/or possible influence from factors such as dilution by ambient snow. These influences were kept to a minimum, but were not completely unavoidable (see methods section for more detail). The order, reaction rate constant (k), half-life, r2 value and normalized threshold value (C*/C0) for each of the labelled compounds are shown in Table 1. The loss rates of all of the compounds, with the exception of heptadecane-d36, were found to be first order. Heptadecane-d36 concentrations remained steady over the 72-h time period. As heptadecane-d36, which did not show any loss, has the highest vapour pressure of all the labelled compounds included in the experiment it is unlikely that any of the compounds were lost due to simple evaporation. The corresponding equations for loss rate are listed in Table 1, and are plotted with the experimental data in Fig. 1, with the curve extrapolated to 96 h. That the concentrations did not decrease to zero may be due to the location of the contaminant in the snow or ice crystals. It has been shown that as liquid water freezes to form snow or ice crystals solutes can be excluded to the quasi-liquid-layer on the surface or other liquid-like-layers throughout the crystals (Sumner & Shepson 1999; Grannas, Bausch et al. 2007). This may result in only a portion of the contaminant being available for loss. The incorporation of a portion of the labelled compounds into the snow crystals in inner liquid-like-layers may be the reason that the loss processes resulted in loss to a threshold/steady-state value. Furthermore, the higher volatility of the lighter heptadecane-d36 relative to docosane-d46, may mean that a portion of the heptadecane-d36 was lost immediately during production while the remaining portion was incorporated in a way so as not to allow for physical loss processes. Volatilization from the snowpack is also a possibility. Herbert et al. found that significant increases in snow density correlated with observed decreases in polychlorinated biphenyl (PCB) concentrations (Herbert et al. 2006). These decreases in snow density were linked with decreases in snow surface area that cause a reduction in the capacity of the snow to retain vapour-sorbed chemicals (Herbert et al. 2006). Given the oxidative nature of the atmosphere above the snowpack in the Arctic during the summer season, reaction with radicals (e.g., OH or OH2 radicals) is also a distinct loss possibility (Domine & Shepson 2002; Dolinova et al. 2006). Possible correlations between these properties and the compounds in this study were investigated, however no correlation between experimental half-lives from this study and vapour pressure or OH radical reaction rates was found. Given the limited data set this does not exclude the possibility of these factors influencing the loss of these compounds from the snow. Insufficient light penetration through the exposed snow is probably not the cause of incomplete degradation or loss as the majority of photochemistry has been documented to take place in the upper 25 cm of the snowpack (Grannas, Jones et al. 2007). In this study the threshold concentrations (C*) occurred after 40–60% of the compound was lost for the alkanes (except heptadecane-d36) and acids, and after 90% of the compound was lost in the case of the PAH.
The half-lives for the labelled compounds were calculated based on the reaction rate constants (Table 1). The half-lives ranged from 9.9 h for hexadecanoic acid-d31 to 17 h for the docosane-d46. For comparison, Klan and collaborators investigated the degradation of various organic compounds in the laboratory, as well as in the field, from artificially made frozen aqueous solutions (Klan et al. 2003; Dolinova et al. 2006). The half-lives reported for the organic compounds in samples containing organic compounds and hydrogen peroxide that were irradiated in the field ranged from 0.5 to 21.3 h for various starting concentrations of naphthalene, anthracene, phenol and benzene (Dolinova et al. 2006), which compares well with our calculations. Additional laboratory studies focused more on persistent organic pollutants, such as chlorophenols, chlorobiphenyls and nitrophenols, not PAHs (Klan et al. 2003). Klan et al. (2003) observed half-lives for eight organic compounds had a much larger range, from 8 min for 2-nitrobenzaldehyde to 75 days for 2-nitrophenol. In both cases the concentrations of the compounds studied ranged from approximately 10−3 to 10−6 mol L−1. These are much greater than the concentrations used in our study (e.g., fluoranthene-d10 was approximately 10−10 mol L−1). In Greenland snow the median concentration from a 3-m snow pit dug in 2005 at Summit was 2.1×10−12 mol L−1 for fluoranthene (von Schneidemesser et al. 2008); Jaffrezo et al. (1994) reported a range of concentrations for fluoranthene ranging from 4.9×10−14 to 3.4×10−12 mol L−1 from a 3-m snow pit near Summit in 1991. For comparison 6.4×10−11 mol L−1 of fluoranthene was measured in snow samples from northern Wisconsin, USA (von Schneidemesser et al. 2008).
Herbert et al. (2006) reported similar findings to those presented here when observing PCB concentrations in snow in the Norwegian Arctic. The initial concentration of PCBs measured after a fresh snowfall were found to decrease by about 75% within the first 96 h (k=0.01 h−1), after which the concentration remained steady for the next 136 h—until the next snowfall (Herbert et al. 2006). As the majority of previous research on the degradation and/or loss of organic compounds in snow and/or ice was conducted on aromatic compounds (Dubowski & Hoffmann 2000; Klan et al. 2003; Dolinova et al. 2006; Kahan & Donaldson 2007; Ram & Anastasio 2009), a comparison of the experimental k-values (and half-lives) are compared with the k-value determined for fluoranthene in this study, shown in Fig. 2. The comparison is divided into experiments in which no co-reactant (specifically H2O2) was added and those which did included H2O2 to photochemically generate OH radicals which then were available to react with the aromatic compounds. As can be seen in Fig. 2, only minor differences are observed between the two. This agrees with the results of Ram & Anastasio (2009), who found that less than 25% of the loss of the PAHs studied was due to indirect degradation due to reaction with the OH radical, and with the lack of correlation between OH radical reaction rates and loss rates for the compounds also observed here, as briefly mentioned earlier. Dolinova et al. (2006) observed an increase in the half-life of the aromatics with decreasing concentrations of H2O2 in the frozen solution; however, the amount of direct versus indirect degradation was not determined. Overall, the loss rates for fluoranthene measured in this study fall within the previously measured values for PAHs, and agree well with the fluoranthene results from Ram & Anastasio (2009). Again, no relationship between loss rates (k) and physical/chemical properties listed in Supplementary Table S1 (e.g., molecular weight, vapour pressure) were found for the compounds presented in this paper, in agreement with results presented by Herbert et al. (2006) and Ram & Anastasio (2009).

Fig. 2
Concentration changes over time, as predicted by comparable reaction rate constants from previous degradation/loss studies applied to the starting concentration of fluoranthene-d10 in this study: (a) rate constants from studies where no additional co-reactants (H2O2) were added; (b) rate constants from studies where H2O2 was added as a source of OH radicals. The following terms are abbreviated: Ram & Anastasio 2009 (R&A); Kahan & Donaldson 2007 (K&D); Dubowski & Hoffmann 2000 (D&H), fluoranthene (FLU), phenanthrene (PHE), pyrene (PYR), anthracene (ANT) and 4-nitrophenol (4NP).
Non-labelled compounds
Non-labelled compounds, including PAHs, alkanes and alkanoic acids (see Table 2 for specific compounds), were incidentally incorporated into the labelled-compound containing snow during the production process due to their presence in the air in the freezer. Their concentration in the snow after production, transport and storage is represented by the control (t=0 h) sets. The general concentration range of the non-labelled compounds in the 0-h exposed snow was 3–12 ng L−1 (PAHs), 100–200 ng L−1 (alkanes) and 200–1200 ng L−1 (alkanoic acids); all reported concentrations were blank corrected. Many of these non-labelled compound concentrations then increased over the 72-h period during exposure on the surface of the ice sheet. This indicates that the downward (or depositional) flux is greater than the loss that is occurring for the compounds measured in this study. To represent the deposition numerically a gross deposition flux (F) component was added to the loss equation to calculate the concentration over time, as shown below:
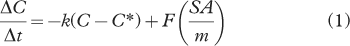
Table 2 Degradation rates and flux values for the non-labelled compounds.
| Compound |
Reaction rate constant used |
Flux (ng hr−1 cm−2) |
| PAHs |
(kg ng−1 hr−1) |
|
| Fluoranthene |
0.068 |
3.7×10−3±2.4×10−3
|
| Phenanthrene |
0.068 |
1.7×10−3±0.97×10−3
|
| Pyrene |
0.068 |
2.110−3±2.2×10−3
|
| Benz[a]anthracene |
0.068 |
1.4×10−3±1.1×10−3
|
| Chrysene |
0.068 |
1.6×10−3±0.99×10−3
|
| Retene |
0.068 |
0.26×10−3±0.14×10−3
|
|
|
|
| n-alkanes |
(hr−1) |
|
| Nonacosane |
0.040 |
7.6×10−3±2.7×10−3
|
| Hentriacontane |
0.040 |
1.9×10−3±0.59×10−3
|
| Dotriacontane |
0.040 |
1.2×10−3±0.45×10−3
|
| Tritriacontane |
0.040 |
1.1×10−3±0.35×10−3
|
| Tetratriacontane |
0.040 |
0.84×10−3±0.19×10−3
|
|
|
|
| n-alkanoic acids |
(hr−1) |
|
| Tetradecanoic acid |
0.068 |
0.026±0.0045 |
| Pentadecanoic acid |
0.068 |
0.019±0.0062 |
| Hexadecanoic acid |
0.070 |
0.090±0.015 |
| Octadecanoic acid |
0.068 |
0.080±0.028 |
| Eicosanoic acid |
0.068 |
0.0040±0.00051 |
| Docosanoic acid |
0.067 |
0.0027±0.0010 |
where C is concentration (ng L−1), t is exposure time (h), C* is the non-reactive threshold concentration (ng L−1), k is the reaction rate constant (per hour), SA is the bulk surface area of the exposed snow (m2) and m is the mass (kg) of the snow being exposed. The deposition fluxes (ng h−1 cm−2) for the non-labelled compounds which were evaluated are listed in Table 2. The standard deviation of the deposition fluxes listed in the table is derived from the variation among the four experiments. The deposition fluxes were calculated based on the 0 and 72 h values only. In the case of experiment 4, where the 0 and 72 h values were from different batches, the average 0 h value from the corresponding batch (batch A) was used. As the deposition is not consistent over time, due to the significant factors of ice rime and fog deposition, an average approach was used. The compounds evaluated include five n-alkanes and six n-alkanoic acids. With the deposition flux and loss rates taken into account, the predicted concentrations were calculated for the non-labelled compounds. For the equation for the predicted concentrations and its derivation, please see the Supplementary File. The deposition of the non-labelled compounds is depicted in Supplementary Fig. S2 for one n-alkane, C32 and one n-alkanoic acid, C14. As a comparison, the corresponding loss rate from the labelled compound is also depicted in Supplementary Figure S2.
The flux values for the PAHs were determined using a simplified equation, based on the average steady-state concentration from the 24- to 72-h period relative to the predicted concentration to be reached by the loss processes. See the Supplementary File for the equation and details. These flux values are also listed in Table 2, and are similar to those of the alkanes. The results are graphically depicted in Supplementary Fig. S3 for non-labelled fluoranthene and chrysene. Overall, the non-labelled PAHs still exhibit a net loss over time relative to their concentration before exposure. In contrast, the non-labelled alkanes and alkanoic acids show an increase in concentration over time relative to their concentration before exposure, indicating that the depositional flux is greater than the loss processes affecting these compounds in the artificially produced snow, assuming that these non-labelled compounds underwent the same loss as their most-similar labelled compound counterparts.
The results presented here show that loss processes (e.g., photochemical degradation, volatilization and reaction with oxidative species) as well as depositional processes (e.g., fog and snowfall) affect the concentration of organic compounds in the snowpack on the order of hours. The methods presented here represent a novel approach to studying post-depositional processing in snow under natural conditions. Further studies on the stability of organic tracer compounds in snow over a longer time period may yield insights as to aerosol deposition rates and their impact on the boundary layer chemistry over permanent snowpacks.
Acknowledgements
This project was funded by the US National Science Foundation (grant no. 0425399). We thank Brandon Shelton at the Wisconsin State Laboratory of Hygiene for sample analysis; Jack Dibb and Casey Anderson of the University of New Hampshire and Gayle Hagler at the Georgia Institute of Technology for planning and field support; Veco Polar Resources staff for servicing Summit Camp and technical support; and New York Air National Guard 109th Airlift Wing for air transit.
References
Cotter
E.S.N.,
Jones
A.E.,
Wolff
E.W.
&
Bauguitte
S.J.B.
2003. What controls photochemical NO and NO2 production from Antarctic snow? Laboratory investigation assessing the wavelength and temperature dependence. Journal of Geophysical Research—Atmospheres 108, article no. 4147, doi: 10.1029/2002JD002602.
[Crossref]
Daly
G.L.
&
Wania
F. 2004.
Simulating the influence of snow on the fate of organic compounds. Environmental Science and Technology 38,
4176–4186.
[Crossref]
Dibb
J.E.
&
Arsenault
M. 2002.
Shouldn't snowpacks be sources of monocarboxylic acids? Atmospheric Environment 36,
2513–2522.
[Crossref]
Dolinova
J.,
Ruzicka
R.,
Kurkova
R.,
Klanova
J.
&
Klan
P. 2006.
Oxidation of aromatic and aliphatic hydrocarbons by OH radicals photochemically generated from H2O2 in ice. Environmental Science and Technology 40,
7668–7674.
[Crossref]
Domine
F.
&
Shepson
P.B. 2002.
Air–snow interactions and atmospheric chemistry. Science 297,
1506–1510.
[Crossref]
Dubowski
Y.
&
Hoffmann
M.R. 2000.
Photochemical transformations in ice: implications for the fate of chemical species. Geophysical Research Letters 27,
3321–3324.
[Crossref]
Grannas
A.M.,
Bausch
A.R.
&
Mahanna
K.M. 2007.
Enhanced aqueous photochemical reaction rates after freezing. Journal of Physical Chemistry A 111,
11043–11049.
[Crossref]
Grannas
A.M.,
Jones
A.E.,
Dibb
J.,
Ammann
M.,
Anastasio
C.,
Beine
H.J.,
Bergin
M.,
Bottenheim
J.,
Boxe
C.S.,
Carver
G.,
Chen
G.,
Crawford
J.H.,
Domine
F.,
Frey
M.M.,
Guzman
M.I.,
Heard
D.E.,
Helmig
D.,
Hoffmann
M.R.,
Honrath
R.E.,
Huey
L.G.,
Hutterli
M.,
Jacobi
H.W.,
Klan
P.,
Lefer
B.,
McConnell
J.,
Plane
J.,
Sander
R.,
Savarino
J.,
Shepson
P.B.,
Simpson
W.R.,
Sodeau
J.R.,
von Glasow
R.,
Weller
R.,
Wolff
E.W.
&
Zhu
T. 2007.
An overview of snow photochemistry: evidence, mechanisms and impacts. Atmospheric Chemistry and Physics 7,
4329–4373.
[Crossref]
Herbert
B.M.J.,
Villa
S.
&
Halsall
C. 2006.
Chemical interactions with snow: understanding the behavior and fate of semi-volatile organic compounds in snow. Ecotoxicology and Environmental Safety 63,
3–16.
[Crossref]
Hutterli
M.A.,
McConnell
J.R.,
Bales
R.C.
&
Stewart
R.W.
2003. Sensitivity of hydrogen peroxide (H2O2) and formaldehyde (HCHO) preservation in snow to changing environmental conditions: implications for ice core records. Journal of Geophysical Research—Atmospheres 108, article no. 4023, doi: 10.1029/2002JD002528.
[Crossref]
Jacobi
H.W.,
Annor
T.
&
Quansah
E. 2006.
Investigation of the photochemical decomposition of nitrate, hydrogen peroxide, and formaldehyde in artificial snow. Journal of Photochemistry and Photobiology A 179,
330–338.
[Crossref]
Jaffrezo
J.L.,
Clain
M.P.
&
Masclet
P. 1994.
Polycyclic aromatic-hydrocarbons in the polar ice of Greenland—geochemical use of these atmospheric tracers. Atmospheric Environment 28,
1139–1145.
[Crossref]
Kahan
T.F.
&
Donaldson
D.J. 2007.
Photolysis of polycyclic aromatic hydrocarbons on water and ice surfaces. Journal of Physical Chemistry A 111,
1277–1285.
[Crossref]
Kawamura
K.,
Kasukabe
H.,
Yasui
O.
&
Barrie
L.A. 1995.
Production of dicarboxylic acids in the Arctic atmosphere at polar sunrise. Geophysical Research Letters 22,
1253–1256.
[Crossref]
Klan
P.
&
Holoubek
I. 2002.
Ice (photo)chemistry. Ice as a medium for long-term (photo)chemical transformations—environmental implications. Chemosphere 46,
1201–1210.
[Crossref]
Klan
P.,
Klanova
J.,
Holoubek
I.
&
Cupr
P. 2003.
Photochemical activity of organic compounds in ice induced by sunlight irradiation: the Svalbard project. Geophysical Research Letters 30,
1313.
[Crossref]
Oros
D.R.
&
Simoneit
B.R.T. 2000.
Identification and emission rates of molecular tracers in coal smoke particulate matter. Fuel 79,
515–536.
[Crossref]
Oros
D.R.
&
Simoneit
B.R.T. 2001.
Identification and emission factors of molecular tracers in organic aerosols from biomass burning. Part 2. Deciduous trees. Applied Geochemistry 16,
1545–1565.
[Crossref]
Patton
G.W.,
Walla
M.D.,
Bidleman
T.F.
&
Barrie
L.A. 1991.
Polycyclic aromatic and organochlorine compounds in the atmosphere of northern Ellesmere Island, Canada. Journal of Geophysical Research—Atmospheres 96,
10867–10877.
[Crossref]
Ram
K.
&
Anastasio
C. 2009.
Photochemistry of phenanthrene, pyrene, and fluoranthene in ice and snow. Atmospheric Environment 43,
2252–2259.
[Crossref]
Rogge
W.F.,
Hildemann
L.M.,
Mazurek
M.A.,
Cass
G.R.
&
Simoneit
B.R.T. 1993.
Sources of fine organic aerosol. 4. Particulate abrasion products from leaf surfaces of urban plants. Environmental Science and Technology 27,
2700–2711.
[Crossref]
Schauer
J.J.
&
Cass
G.R. 2000.
Source apportionment of wintertime gas-phase and particle-phase air pollutants using organic compounds as tracers. Environmental Science and Technology 34,
1821–1832.
[Crossref]
Schauer
J.J.,
Rogge
W.F.,
Hildemann
L.M.,
Mazurek
M.A.
&
Cass
G.R. 1996.
Source apportionment of airborne particulate matter using organic compounds as tracers. Atmospheric Environment 30,
3837–3855.
[Crossref]
Sheesley
R.J.,
Schauer
J.J.,
Bean
E.
&
Kenski
D. 2004.
Trends in secondary organic aerosol at a remote site in Michigan's Upper Peninsula. Environmental Science and Technology 38,
6491–6500.
[Crossref]
Sheesley
R.J.,
Schauer
J.J.,
Zheng
M.
&
Wang
B. 2007.
Sensitivity of molecular marker-based CMB models to biomass burning source profiles. Atmospheric Environment 41,
9050–9063.
[Crossref]
Stone
E.A.,
Snyder
D.C.,
Sheesley
R.J.,
Sullivan
A.P.,
Weber
R.J.
&
Schauer
J.J. 2008.
Source apportionment of fine organic aerosol in Mexico City during the MILAGRO experiment 2006. Atmospheric Chemistry and Physics 8,
1249–1259.
[Crossref]
Sumner
A.L.
&
Shepson
P.B. 1999.
Snowpack production of formaldehyde and its effect on the Arctic troposphere. Nature 398,
230–233.
[Crossref]
von Schneidemesser
E.,
Schauer
J.J.,
Hagler
G.W.
&
Bergin
M.H. 2009.
Concentrations and sources of carbonaceous aerosol in the atmosphere of Summit, Greenland. Atmospheric Environment 43,
4155–4162.
[Crossref]
von Schneidemesser
E.,
Schauer
J.J.,
Shafer
M.M.,
Hagler
G.S.W.,
Bergin
M.H.
&
Steig
E.J. 2008.
A method for the analysis of ultra-trace levels of semi-volatile and non-volatile organic compounds in snow and application to a Greenland snow pit. Polar Science 2,
251–266.
[Crossref]